实验
实验
分离物质
色谱
其他
薄层层析
样品要求:溶液,对254nm或3__nm的光有吸收,或产生荧光。
实验装置
结构
原理
同柱层析1
原理
样品在附着在固定相上,被流动相洗脱到流动相中又再次附着。如此往复,在固定项附着强的,与流动相溶解差的,被流动相冲出的距离短。
与薄层层析相似,均基于化合物在不同相之间的亲和力差异。
通常,薄层层析的固体相位硅脂,并附有荧光剂。当使用紫外灯照射时,硅脂板发出荧光,被样品覆盖处无荧光,即可得知样品位置。紫外光通常取254nm,因为大\pi键在这个波长上多有吸收。
操作
-
预处理
- 如果样品是固体,用毛细管轻戳固体样品,确保毛细管内有固体。将毛细管在固体粉末上轻戳(过重毛细管断裂),然后吸入少量溶剂
-
取样
将毛细管尖端置于液面下,经毛细作用液体吸入毛细管。
-
点板
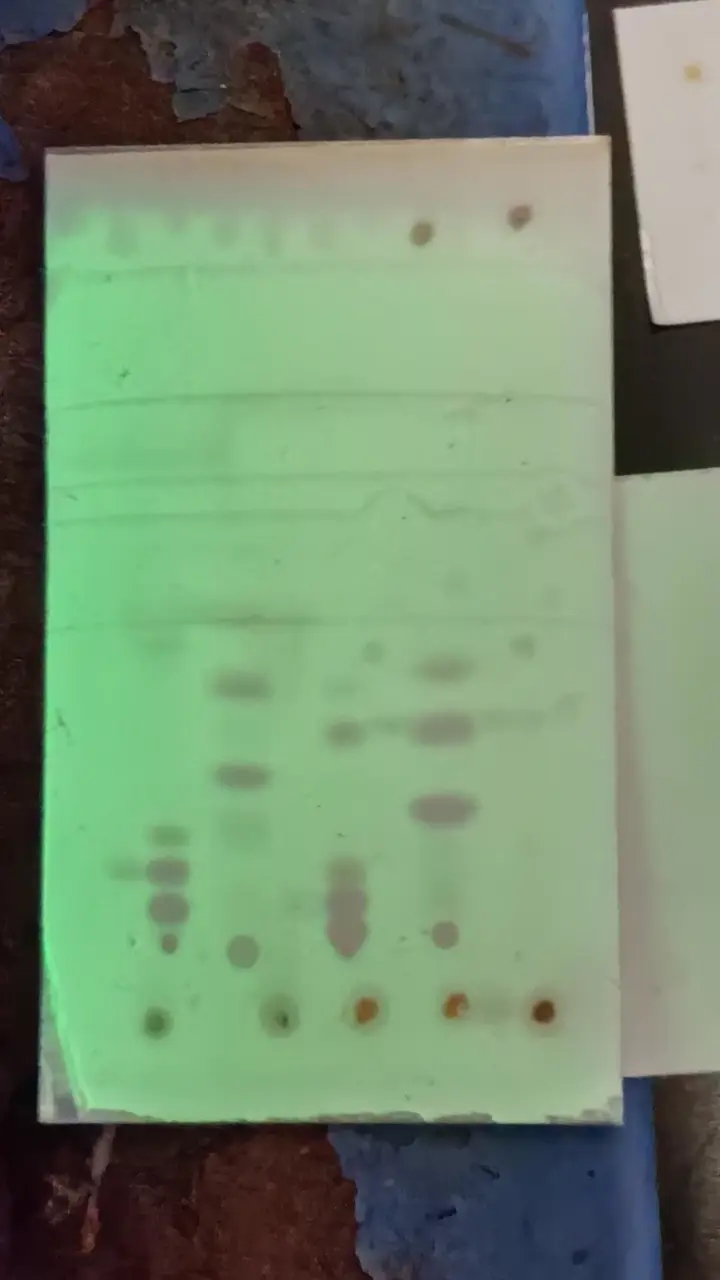
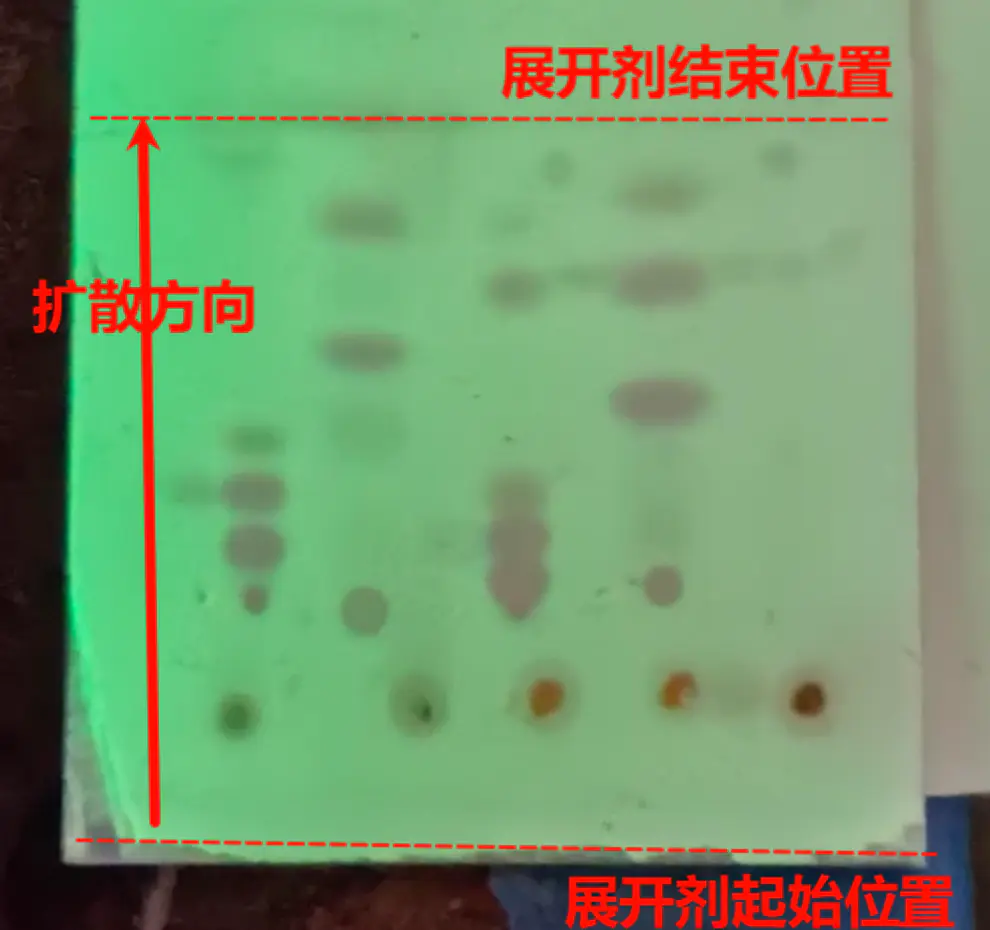
在紫外光照射下,将毛细管垂直轻点在板边缘约1~2 cm处,如未观察到荧光被遮挡(溶液过稀),保持中心不变多次点板。
通常,需要将多个样品对比(例如,将反应过程中的溶液样品与反应物对比,确定反应是否完全),注意保证样品中心在同一直线上,且平行于板边缘。

-
展开
如果只需要知道样品中吸收254nm物质是否出现,此处可跳过直接观察即可。常用于柱层析1
配置展开剂,根据样品极性选择合适的展开剂,目的是展开剂与样品极性相近。展开剂常用乙酸乙酯和石油醚的混合溶液。
初次展开,先尝试极性较小的展开剂,避免样品随展开剂直接扩散到板边缘甚至板外。注意展开剂量较少,避免覆盖样品点,将样品完全溶解。
-
分析

以左一左二为例,假定左一为反应物,左二为反应过程中样品。左二除了与左一相同位置出现标记,还有上下两个。可以认为反应为完全,如果已知最远的是催化剂,则生成物为最近的。
-
柱层析
样品要求:干燥的固体,对254nm吸收
实验装置

结构
-
固定相:一种与样品有亲和力(吸附性)的物质,且不溶于流动相。通常为固体。
- 硅胶,极性强,常用于极性样品
- 氯化铝,非极性。
-
流动相:一种样品的(最好是产物的)良好溶剂,且固定相不溶解。为了检验方便,通常选择对254nm不吸收的溶剂。又称洗脱剂。
-
石英砂:覆盖在样品上方,防止洗脱剂将样品冲散
原理
样品在附着在固定相上,被流动相洗脱到流动相中又再次附着。如此往复,在固定项附着强的,与流动相溶解差的,被流动相冲出的距离短。
与薄层层析相似,均基于化合物在不同相之间的亲和力差异。
操作
-
预处理
若待测样品为溶液,加入样品2~3倍质量的硅脂干燥。一定保证固体为粉末状。
-
加固定相(以硅脂为例,使用湿法装柱)
- 在柱底塞入少量棉花,避免硅胶流出,但勿塞得过紧,以免影响流速。
- 将硅脂用洗脱剂(二氯甲烷或石油醚)溶解后,边搅拌边加入柱子中,避免分层。
- 加入到较满后,加压或常压等待溶剂流下,硅脂均匀地堆积在柱子中。
- 重复上述操作直到硅脂高度约15cm,如大于15cm过柱速度较慢,但精度无影响。
-
润洗
- 加入洗脱剂充分冲洗柱床,移除杂质并使其稳定。
-
加样品
将样品均匀地铺在固定相上方,如不均匀可轻敲柱子。
-
加石英砂
为避免流动相冲击样品使得样品表面不平,加入不溶于流动相的石英砂约2cm厚。
-
洗脱
-
后处理
- 收集含有目标物的洗脱液,使用旋转蒸发仪(旋蒸)除去溶剂,即得目标化合物。
-
液相色谱
样品:溶液,紫外光谱有与展开剂相比有显著不同的吸收峰。
实验装置
原理
同柱层析,
液相色谱内置了可变波长的紫外灯,可以通过检测分离出的物质对特定hv的吸收强度确定是否有物质流出、流出物质的多少。故吸收强度对时间积分正比于物质的量。
相同物质,相同溶剂和展开剂下,流速一定,冲洗时长一定相同。所以可以通过对已知物质进行液相色谱,与未知溶液对比,可以确定未知溶液某物质占比。
通常,作 吸光度-时间 曲线用来分析数据,例如
结构
注射口,液相色谱仪,柱子。
根据极性不同选择不同的展开剂和柱子。
操作
-
初始化
启动液相色谱仪,启动紫外灯,通过其他色谱方法确定使用的紫外光波长。
-
安装柱子并润洗
-
挑选合适的柱子和展开剂
-
安装柱子
-
打开液体阀门,用较快的流速润洗管道并排除气泡两到三次
-
关闭液体阀门,用较快的流速润洗柱子
-
观察hv吸收强度,等到吸收强度稳定在一个常数时,认为管道初步润洗完成。
如果吸收强度忽然很大,可能是气泡或上次使用了极性较强的展开剂。
-
改用目标流速(通常0.8 mL/min)润洗柱子,完成润洗判定同上。
-
设定当前吸光强度为0
-
-
吸取溶液(该操作可与润洗柱子1同时进行)
- 查看进液口,确定设备接受的溶液体积,下以\mathrm{20\,\mu L}为例。
- 使用前,将针头在丙醇溶液(展开剂)中润洗3次。
- 匀速快速地吸取待测溶液,略大于设备接受体积,如\mathrm{25\,\mu L}.
- 如管内有气泡,将针管向上,弹击针管使气泡在顶部。
- 将溶液推到约\mathrm{17\,\mu L}, 排除气泡。推出时用吸水纸包裹针头。
-
推入溶液
- 将针管插入进液口
- 快速将阀门打开到头,快速推入溶液,快速关闭阀门(避免气泡进入)
- 拔出针管,在丙醇溶液(展开剂)中润洗3次。
-
观察结果
↩
-
- 改用目标流速(通常0.8 mL/min)润洗柱子,完成润洗判定同上。 ↩
旋蒸
目的:从高沸点溶液中分离低沸点物质(或低沸点溶液分离高沸点物质)
原理
--
操作
-
预处理:
如果溶剂和产物沸点差距小,可尝试先进行萃取。例如用二氯甲烷将反应物从溶剂中萃取到有机相,实现了将溶剂更换为低沸点的二氯甲烷。
-
开始旋蒸
- 打开循环水压缩机(冷凝水制冷和水泵装置,可以使用流动水源代替)
- 将样品倒入烧瓶,不超过1/3,如过多分多次。
- 将烧瓶连接到防爆管上,握住烧瓶避免烧瓶掉下。
- 关闭气阀
- 打开真空泵,待瓶内气压下到0.3~0.4 atm时,松开烧瓶并开启旋蒸仪。
-
处理爆沸
如果旋蒸途中发生爆沸,处理方法
- 立即打开气阀,使大气进入旋蒸体系。
- 在爆沸停止后快速关闭气阀,避免压力差过小烧瓶掉下。
考虑以下方法减少爆沸
- 降低旋蒸温度
- 降低转速
- 加碎瓷片
-
结束旋蒸
- 停止旋转
- 手握烧瓶,打开气阀放气。
- 待气压恢复到接近1 atm时取下烧瓶
- 根据实际情况:关闭真空泵,关闭循环水压缩机。
↩
-
萃取
样品:根据样品在极性溶剂和非极性溶剂中的溶解性不同分离
实验装置
原理
样品中不同物质极性不同,将样品加入极性小的有机溶剂(萃取剂)和水的体系中,进而分离物质。
萃取剂常选:乙酸乙酯、异丙醇等。
如果希望将溶剂溶到水层中,产物溶解到有机溶剂中,注意考虑溶剂和萃取剂是否相溶。
结构

操作
-
加入样品和萃取剂
- 将样品加入梨形分液漏斗中
- 加入萃取剂
- 加入饱和食盐水(进一步增强水相极性,破坏乳化)
-
摇匀
-
塞紧塞子
-
取下任意方式摇匀
-
每隔一会将放液口朝无人处,上扬漏斗,缓慢打开放液口放出气体
朝无人处:有液体冲出的风险
-
重复上述操作数次
-
-
静置
静置直到溶液分层,界限分明。
-
放液
-
重复萃取
(以产物在有机相为例)第一次的水层需要再次萃取。
-
处理乳化现象
如果观察到界限较为分明,但水相(或有机相)仍有一些不融的沉淀或液体(如下图),一般是发生了乳化现象。

潜在原因
- 因某些原因产生表面活性物质。
- 形成乳浊液
- 形成胶体
- etc.
处理方法
- 延长静止时间
- 加入饱和食盐水
- 分出有机相(水相),再次萃取水相(有机相) ↩
-
